Researchers in the Stephenson School of Biomedical Engineering, Gallogly College of Engineering at the University of Oklahoma, have developed a framework published in Science Advances that solves the challenge of bridging experimental and computer sciences to better predict peptide structures. Peptide-based materials have been used in energy, security and health fields for the past two decades.
Handan Acar, Ph.D., the Peggy and Charles Stephenson Assistant Professor of Biomedical Engineering at OU, teamed up with Andrew White, Ph.D., an associate professor of chemical engineering at the University of Rochester, to introduce a new strategy to study fundamentals of molecular engineering. Seren Hamsici, a doctoral student in Acar’s lab, is the first author of the study.
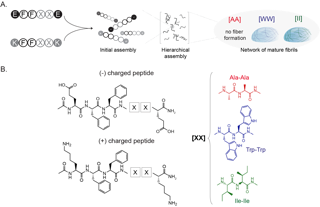
Proteins are responsible for the structure, function and regulation of the body’s organs and tissues. They are formed by amino acids and come together in different interactions, called intermolecular interactions, that are essential to how proteins perform different roles in the body. When these protein interactions behave abnormally, medical issues result, such as when they clump together to form plaques in the brain that leads to Alzheimer’s Disease.
“In the peptide-engineering field, the general approach is to take those natural proteins and make incremental changes to identify the properties of the end aggregated products, and then find an application for which the identified properties would be useful,” Acar said. “However, there are more than 500 natural and unnatural amino acids. Especially when you consider the size of the peptides, this approach is just not practical."
Machine learning has great potential to counter this challenge, but Acar says the complex way peptides assemble and disassemble has prevented artificial intelligence methods from being effective so far.
“Clearly, computational methods, such as machine learning, are necessary,” she said. “Yet, the peptide aggregation is very complex. It is currently not possible to identify the effects of individual amino acids with computational methods.”
To counter those challenges, the research team came up with a new approach. They developed a framework that would help bridge materials science and engineering research with computational science to lay the groundwork for artificial intelligence and machine learning advancements.