
AlphaFold 2
AlphaFold 2 (AF2) is a GPU-accelerated machine learning system used to predict 3D protein structures. It includes two models: one for monomers and another for multimer complexes. As with any computational method, AF2 predictions should be interpreted with care and validated using experimental data or complementary computational tools. Refer to the AF2 documentation for details on its limitations.
References
Usage
To run the default installed version, load the module:
module load AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1
Set paths to AlphaFold databases and installation directory:
export ALPHAFOLD_DATA_DIR=/opt/oscer/software-el9/AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1/data
export ALPHAFOLD_DIR=/opt/oscer/software-el9/AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1
Run the AlphaFold script:
python $ALPHAFOLD_DIR/bin/run_alphafold.py \
--fasta_paths=/path/to/your/sequence.fasta \
--max_template_date=2022-01-01 \
--model_preset=monomer \
--models_to_relax=none \
--output_dir=/path/to/output/directory
Refer to the AF2 GitHub repository for a full list of available flags.
Example Job
Monomer Structure Prediction
Here is an example of a job using 4 CPU cores for the multisequence alignment (MSA) step and 1 GPU for model inference.
- Test case: small ultra-red fluorescent protein (140 amino acids; identifier 7UQA).
- Runtime: ~75minutes.
- Memory usage: ~42 GB
Note: Update the #SBATCH --chdir directive to point to your own working directory
#!/bin/bash
#SBATCH --partition=gpu
#SBATCH --container=el9hw
#SBATCH --gres=gpu:1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
#SBATCH --mem=48GB
#SBATCH --time=03:00:00
#SBATCH --job-name=af2_test
#SBATCH --output=af2_%j.stdout.txt
#SBATCH --error=af2_%j.stderr.txt
#SBATCH --chdir=/path/to/working/directory
# Load necessary modules
module purge
module load AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1
# Set path to AlphaFold databases
export ALPHAFOLD_DATA_DIR=/opt/oscer/software-el9/AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1/data
# Set the path to the AlphaFold installation directory
export ALPHAFOLD_DIR=/opt/oscer/software-el9/AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1/
# Run Alphafold2
echo "Job started at: $(date)"
python $ALPHAFOLD_DIR/bin/run_alphafold.py \
--fasta_paths=$ALPHAFOLD_DIR/examples/monomer/7UQA.fasta \
--max_template_date=2023-01-01 \
--model_preset=monomer \
--models_to_relax=none \
--output_dir=$SLURM_CHDIR
echo "Job finished at: $(date)"
Important Notes
- AF2 first runs multithreaded MSA, then runs model inference on the GPU. Increasing CPU cores beyond 4 provides minimal performance improvement.
- Memory usage can be high (e.g., ~42 GB for a 140 AA protein) and scales with protein size.
- FASTA file input: You must change the template script to point to your FASTA file: --fasta_paths=/path/to/your/sequence.fasta
- Template date: Please note you should change the --max_template_date to suit your analysis
- Example inputs are available at: /opt/oscer/software/AlphaFold/2.3.2-foss-2023a-CUDA-12.1.1/examples
Output Files
AF2 will create a directory with the jobID under your specified output directory. This will contain:
- Predicted structures in PDB format. The ranked_0.pdb file corresponds to the model with the highest confidence.
- Python pickle files (.pkl). Unlike the PDB files, these are not reordered into ranked outputs. They store extensive model datathat can be explored or visualized using Python.
- Confidence metrics, including pLDDT and PAE scores.
- Log files documenting the run.
- MSA visualization files.
Bechmarks
To evaluate AlphaFold2 runtimes, two individual proteins were processed across all available GPUs in the sooner_gpu_test partition. The test cases included a small ultra-red fluorescent protein (140 amino acids; PDB ID: 7UQA) and carotenoid oxygenase 1 (526 amino acids; PDB ID: 5U8X). All runs used default settings, without relaxation, and a fixed --max_template_date=2023-01-01.
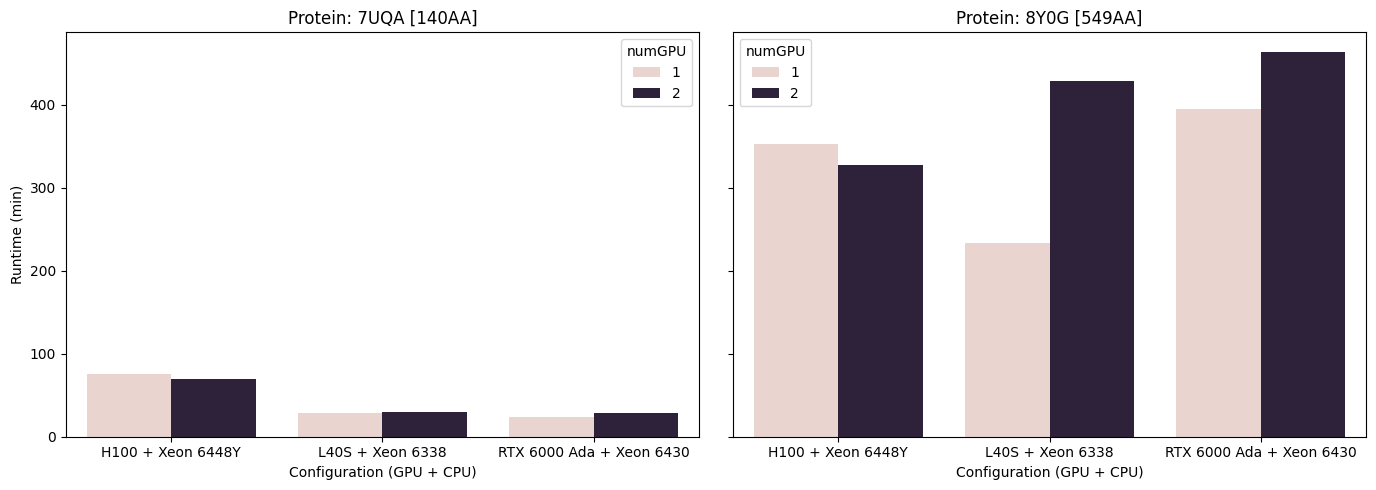
Figure 1. AlphaFold runtimes for two proteins using 4 CPUs and 48 GB (7UQA) and 60 GB (5U8X) of memory. The 7UQA protein is a small ultra-red fluorescent protein (140 aa), while 5U8X corresponds to carotenoid oxygenase 1 (526 aa). Note that (1) dual-GPU configurations without NVLink (L40S and RTX 6000) performed worse than their single-GPU counterparts, in contrast to the H100 configuration (2) for smaller inference workloads, such as the 7UQA protein, performance differences between GPUs were minimal (3) a single L40S paired with a Xeon 6338 CPU matched or outperformed the other tested configurations.
Overall, the AlphaFold2 pipeline shows suboptimal resource use, with most computation dominated by CPU-intensive MSA generation. Separating pipeline stages into appropriately resourced jobs may improve performance, which is in the works.
Issues
If you encounter an error such as:
RuntimeError: HHSearch failed
it is most likely due to insufficient memory allocation in your job script, and you should request more memory.
